







Table of contents
- Installing and starting VIZARD
- Loading data
- Filtering
- View
- Sorting
- Clustering
- Navigating and cleaning data
- Regulatory motif search
- Tools
- Saving results
Installing and starting VIZARD
VIZARD will run on any Java-enabled platform supporting Java 2 (JRE 1.2.2 and later). Download the installation program for your operating system and start it. It will guide you through the installation process. If the Java 2 Runtime Environment (JRE) is not included in your distribution, you can download the JRE for your platform from the Sun Microsystems web site. Since MacOS does not support Java 2, the program will not run on Mac. After successful installation you will find a new VIZARD folder and icon in this folder. If you install the program on a Windows computer, you will also find VIZARD's icon in the Programs menu. You can start VIZARD using this icon.
Loading data
The most convenient way to create an input dataset file is to use a spreadsheet
program such as MS Excel and save the file in a tab-separated format (Save As...
Text (Tab delimited)"). You can find the sample dataset, SampleDataSet.txt
file, in the VIZARD installation folder under the data directory.
Another way to load the sample dataset is to use the Help -> Load
Sample Data Set menu. After creating the input data set file launch VIZARD
and open the file using the File -> Open menu or the ![]() Open button the toolbar. VIZARD
expects the following tab-separated file format:
Open button the toolbar. VIZARD
expects the following tab-separated file format:
- Probe set ID (number or text)
- Description (text)
- First (control) experiment Absolute Call (optional, P, A, or M; stands for "Present", "Absent", or "Marginal", respectively)
- First (control) experiment expression level (number; Average Difference of the Affymetrix GeneChip® array)
- Second experiment Absolute Call (optional)
- Second experiment expression level
- And so on...
For example,
17362_s_at glucosyltransferase P 182 P 90 P 39 A -2or
17362_s_at glucosyltransferase 182 90 39 -2
Filtering
Go to the Analyze -> Filter microarray data menu or hit the ![]() Filter button
and change or accept default filtering criteria. You should see:
Filter button
and change or accept default filtering criteria. You should see:

The following parameters are used:
- Noise Multiplier (QMult) = 2.8 or 2.1 for arrays with 24 or 50 micrometers feature size, respectively. It is 2.8 for Arabidopsis 8k GeneChip®.
- Maximal Raw Noise (RawQ) = maximal RawQ value between all compared experiments.
- Fold Change Threshold = fold change threshold for the gene expression level. Normally, it should be more than or equal to 2.
- Minimal Expression Level Difference = minimal difference between expression level values of one gene in all compared experiments (i.e. minimal difference between Average Differences). It should be more than or equal to QMult * RawQ. This parameter is used to filter out genes which pass the Fold Change Threshold, but their expression levels were too close to the background/noise level to be considered as really differentially expressed.
- Number of Present Transcripts = Minimal number of Absolute Call "P" values for one gene in all compared experiments. Normally, it should be more than or equal to 1. It is used to filter out genes, which have "A" (Absent) or "M" (Marginal) Absolute Calls, because of either (i) their expression levels were too close to the background/noise level, or (ii) cross-hybridization (or other adverse conditions) affected reliability of data for these genes in a particular experiment. Therefore, if, for example, you have four experiments and you want to filter out all genes that had "A" or "M" Absolute Calls in any of these experiments, you should set the Number of Present Transcripts to 4. Be careful, if you do this, some of the removed genes could have "A" or "M" Absolute Calls because of down-regulation to the background/noise level. This parameter will not be available if your input file does not contain Absolute Call values.
- Use Exclusion Experiment? is an optional but useful parameter when you want to filter out genes either (i) showing significant expression changes (usually, above 2-fold) in a mock experiment, or (ii) showing expression changes below a given fold change threshold in a selected experiment of interest (for example, at 30 min time point in time series), or (iii) showing expression changes above a given fold change threshold in all experiments except a selected experiment of interest (for example, to see what genes were differentially expressed only at 30 min time point). If you check Yes, you will be presented with the following additional filtering parameters:
- Use This Experiment as Exclusion Experiment = Exclusion Experiment number. Can be the number of any experiment of interest except of the first (control) experiment (i.e., valid values are from 2 to the total number of experiments in the data set).
- EE Fold Change Threshold (EE FCT) = fold change threshold for the Exclusion Experiment.
- Remove Genes In Which: = Exclusion Experiment filtering category. If you choose the EE FC > EE FCT, genes showing expression changes above EE FCT in the Exclusion Experiment will be filtered out from all experiments. Often, the Exclusion Experiment is an additional control or mock experiment, and EE FCT is usually less than or equal to 2 in this case. If you choose EE FC < FCT, genes showing expression changes below Fold Change Threshold in the Exclusion Experiment will be filtered out from all experiments. This is useful, for example, when you are interested in genes showing significant differential expression in a particular experiment. Note that Fold Change Threshold is used in this case, not EE FCT. Finally, if you choose Other FC > EE FCT, genes showing expression changes above EE FCT in ALL EXPERIMENTS OTHER THAN the Exclusion Experiment will be filtered out. This is useful, for example, when you are interested in genes that show significant expression changes in one particular experiment only (which in this case is Exclusion Experiment)
After clicking on the OK button, you will typically see an image similar to the following:

First two columns ("Id" and "Description") are self-explanatory. Fold Change values in the rest of the columns represent expression level changes. They are relative to the first data column ("Exp1"), which is normalized to 1 (usually, it is a control experiment). Increase in mRNA abundance (up-regulation) is shown in red, decrease (down-regulation) in green.
View
Color encoding for up- and down-regulated genes can be customized via the View -> Change expression colors... menu. For example, down-regulation can be shown in blue.
 Zoom in
and
Zoom in
and  Zoom out
commands of the View menu (and the corresponding buttons on the toolbar)
change color encoding, so that low fold change values become more visible or less visible,
respectively.
Zoom out
commands of the View menu (and the corresponding buttons on the toolbar)
change color encoding, so that low fold change values become more visible or less visible,
respectively.
"Look and feel" (Metal, Motif or Windows) of the program can be changed to a user preference via the View -> Change look & feel menu. The Windows look & feel option will be available only on Windows computers.
Sorting
After filtering microarray data you can change default sorting order via
the Analyze -> Sort filtered data menu or the ![]() Sort button on the toolbar.
The Sort menu has two options:
Sort button on the toolbar.
The Sort menu has two options:
- Absolute - all data will be sorted in the following order: the greatest gene expression changes in all compared experiments will show up at the top, the lowest at the bottom. Expression changes are "absolute", i.e. they are sorted regardless of their "+" (increased expression level, up-regulation) or "-" (decreased expression level, down-regulation) signs. This is useful when you want to see at the top genes that showed the greatest fold changes regardless of either they were up- or down-regulated.
- Relative - data will be sorted in the following order: the largest gene expression changes with the "+" sign will show up at the top, the lowest "+" and "-" changes in the middle, and the largest changes with the "-" sign at the bottom. This is useful when you want to see at the top up-regulated genes with the greatest fold changes.
Clustering
Genes can be clustered (or grouped together)
via the Analyze -> Group/cluster genes with similar or opposite expression patterns
menu or the ![]() Cluster button on the toolbar.
Clustering can be done using the following three options:
Cluster button on the toolbar.
Clustering can be done using the following three options:
- Positive Pearson Correlation - genes with similar expression patterns will be clustered using the Pearson Product Moment Correlation (called Pearson's correlation for short) which is the most common measure of correlation. Gene clusters with the strongest correlation will appear at the top, with the weakest - at the bottom.
- Negative Pearson Correlation - genes with negatively correlated expression patterns will be clustered pairwise using Pearson's correlation. This is useful when you want to find out what gene had an opposite expression pattern compared to a selected gene of interest.
- Submit clustering job to the European Bioinformatics Institute (EBI) EPCLUST. In this case VIZARD will submit your filtered data to the EPCLUST, open a system browser (MS Internet Explorer or Netscape) you will typically see an image similar to the following:

Hit the Select the corresponding experiments button. On the next screen, hit either the Select the data! button, to submit data for all experiments (columns), or the Show column-wise information, allow to manually select certain columns button, to select which experiments include for clustering. On the next screen, change or accept default parameters, and click on the GO! button corresponding to Hierarchical clustering or K-means.
Navigating and cleaning data
To find any particular gene of interest, either go to the Search -> Find
menu or click on the  Find button
on the toolbar.
Find button
on the toolbar.
Double click on the gene of interest and data view will change from the Fold Change to the Expression Level where you can see Average Differences and Absolute Calls (if available).
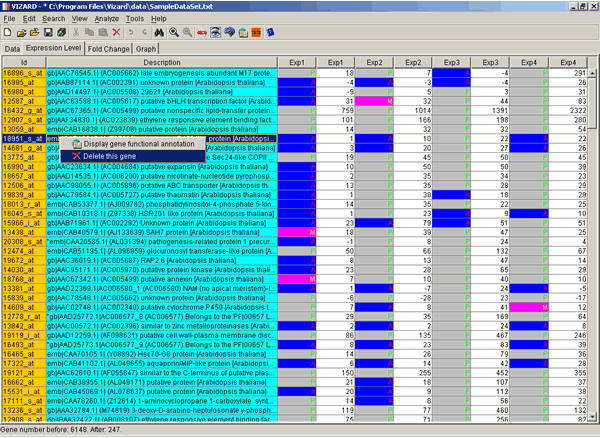
Double click again, and you will be returned to the Fold Change view. Right-click on any gene of interest with
your mouse and you will see a pop-up menu with two options:  Display gene functional
annotation and
Display gene functional
annotation and  Delete this gene. If you choose the
Display gene functional annotation option, VIZARD will launch a system
browser and will display all gene information available at the MATDB in the browser window.
Delete this gene. If you choose the
Display gene functional annotation option, VIZARD will launch a system
browser and will display all gene information available at the MATDB in the browser window.
Usually, to obtain reliable results, the data requires cleaning.
If you are not confident in expression level changes of any particular
gene (for example, when all compared experiments have "A" or "M" Absolute Calls for the gene),
you can delete that gene from a data set.
To do this, select the gene in the Expression Level view or in the
Fold Change view and use either the Edit -> Delete menu or the Delete
button on the toolbar, or click on the gene with the right mouse button and choose the Delete this gene
command from the pop-up menu.
To see graphic representation of a gene expression profile, click on the Graph tab.

If you click on the Data tab, you can use text editor capabilities of the
program such as  Cut,
Cut,  Copy,
Copy,
 Paste, Delete,
Paste, Delete,
 Undo,
Undo,  Redo, and Select All.
This is useful, for example, when one wants to remove gene clusters that he or she is not interested in.
Redo, and Select All.
This is useful, for example, when one wants to remove gene clusters that he or she is not interested in.
Regulatory motif search
Regulatory motif searches are done via integration with AlignACE
software created at the Church Lab, Harvard Medical School, Harvard University. At present, AlignACE program is one of
the most popular and advanced motif discovery programs used in microarray data analysis. VIZARD extracts and formats upstream sequences of genes
either left after filtering or belonging to any particular cluster of interest, and then submits them as input to
AlignACE. VIZARD also automatically computes GC-content of these upstream sequences and provides it as
GC-background parameter for AlignACE. Since AlignACE is currently supported only on the Linux
platform, VIZARD can invoke AlignACE using either client-server approach (for example, when a user
works at the Windows computer) or locally, when both programs are installed on the same Linux computer. If you have
a Linux machine install AlignACE program, which can be downloaded here.
Next, filter and, optionally, cluster your data. Then either go to the Analyze -> Find shared motifs in upstream sequences menu
or hit the ![]() AlignACE button. You will be presented with a dialog for the AlignACE program.
Enter full file path to the AlignACE program file (e.g., /home/nick/align/AlignACE) and
select either the Use upstream sequences of genes left after filtering option or
copy and paste probe set IDs and their descriptions from a chosen EPCLUST cluster into the corresponding
text area.
AlignACE button. You will be presented with a dialog for the AlignACE program.
Enter full file path to the AlignACE program file (e.g., /home/nick/align/AlignACE) and
select either the Use upstream sequences of genes left after filtering option or
copy and paste probe set IDs and their descriptions from a chosen EPCLUST cluster into the corresponding
text area.
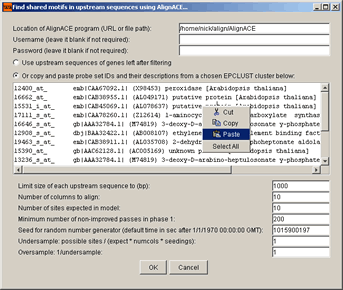

After clicking the OK button, the following window will appear:

Once VIZARD receives results from AlignACE, it can process the results, display and save motif statistics. It can also submit the motif search results to the EBI SEQUENCE LOGO web tool for sequence logo generation. After VIZARD has downloaded a generated image file from the EBI site, you can save it on the local hard disk as a GIF file.

If you have both a Windows and a Linux computer, you can set up the Linux computer as a server, install AlignACE on it and tell VIZARD where to send requests. A source code of a JSP (Java Server Page) for networking with VIZARD and AlignACE can be found here.
Tools
The Tools menu has the following commands:
- Download up-to-date annotations for all genes will download functional annotations from the MATDB at MIPS and save them locally. This will be done for all genes which are present in the GeneDescription.txt (located under the data directory in the VIZARD installation folder) and which have protein entry codes or AGI identifiers (e.g. At1g78310). VIZARD uses the AGI identifiers extracted from the Arabidopsis GeneChip® index file created at the Schroeder Lab, University of California, San Diego. The GeneDescription.txt file should be updated with new downloaded data.
- Update annotations for genes left after filtering will update gene functional annotations using the GeneDescription.txt file. If you do not save your data file after this command, all updates will be lost (but you can always update annotations later).
- Download upstream sequences for all genes will download upstream sequences from the MATDB at MIPS and save them locally. This will be done for all genes which are present in the GeneDescription.txt file and which have protein entry codes. Size of the downloaded upstream sequences is set to 1000 base pairs. If sequences with size less than 1 kbp have been downloaded, their IDs and size will be saved in the VIZARD log file.
- Extract upstream sequences for genes left after filtering will extract upstream sequences using the upstreamAll.seq file under the data directory in the VIZARD installation folder. Upstream sequences will be saved in the FASTA format suitable for the majority of promoter- and motif-finding programs.
- Sequence Utility will perform simple sequence transformation tasks such as make complement, reverse and reverse complement sequences from the input DNA sequence. It will remove any alpha-numerical characters except 'A', 'T', 'G', and 'C'.
Saving results
- If you click on the File -> Save or the
 Save button
on the toolbar, filtering/sorting/clustering results will be saved with
the same name as of the opened file except an extra *.xls extension will be
added (as a convenience feature for fast, double click opening of the saved
file in MS Excel). The saved file can be later opened in VIZARD, any text editor or MS Excel.
An original input dataset file used for filtering/sorting/clustering
will remain intact, unless you use the Save As command and save
the file under the same name as the input data file.
Save button
on the toolbar, filtering/sorting/clustering results will be saved with
the same name as of the opened file except an extra *.xls extension will be
added (as a convenience feature for fast, double click opening of the saved
file in MS Excel). The saved file can be later opened in VIZARD, any text editor or MS Excel.
An original input dataset file used for filtering/sorting/clustering
will remain intact, unless you use the Save As command and save
the file under the same name as the input data file.
- To save under a different file name or with a different file extension or to overwrite the original input file, use the File -> Save As.
- To save fold changes, click on the File -> Save Fold Changes .
- To save filtering results in file formats recognized by supported clustering programs, use File -> Export to... -> [Clustering Program]. Currently VIZARD supports file formats for MIT/Whitehead GeneCluster, B. Dysvik's J-Express, and M. Eisen's Cluster. IMPORTANT NOTE: Make sure that a name under which your file will be saved when exporting to MIT/Whitehead GeneCluster has a "*.res" file extension, otherwise, you will not be able to open the exported file in GeneCluster.